
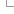
BioMart
- Setting your Filters
- Setting your Attributes (outputs)
- Downloading your Output
- Accessing data using R
BioMart is a simple, web-based tool for mining data from the WormBase ParaSite database.
There are two main parts to using BioMart, which can be navigated between in the menu on the left of the tool:
- Filters: defines the data you would like to include as input for your query
- Attributes: defines the data types you want as output
1) Setting your Filters
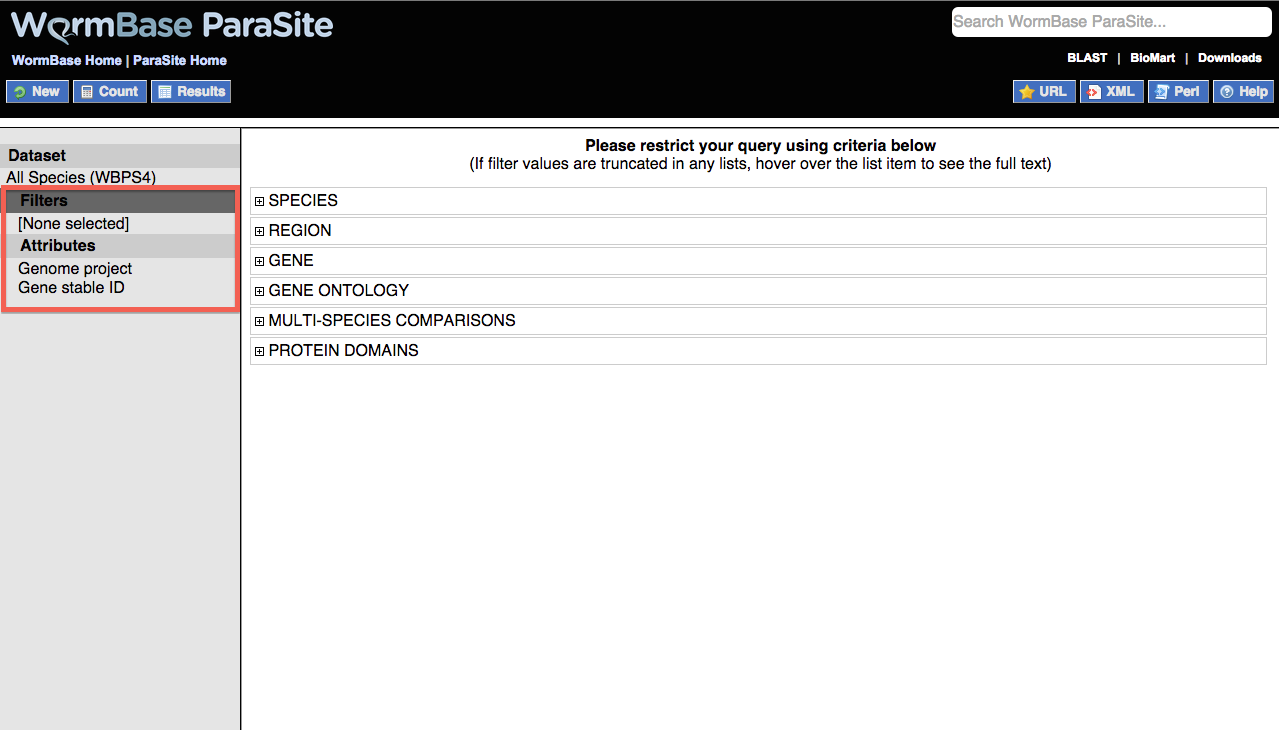
Filters are shown as a list in which each of the items can be expanded by clicking on the + sign:
The following filters are available:
- SPECIES: Use this filter to select either individual genomes or nematode clades. Multiple genomes can be selected by holding down the ctrl key or the option key on a Mac.
- REGION: Restrict to a particular genomic region. Note that this filter should only be used where a single genome has been selected, as it is possible that a particular region is present in multiple genomes.
If start/end co-ordinates are being specified, a scaffold or chromosome id is always required.
Where multiple regions are specified, the format is 'Scaffold/Chr:Start:End:Strand' e.g. AG00032:411187:446321:1. If no strand is specified, both strands are selected. Regions should be separated by a comma or new line.
- GENE: Specify a list of genes with WormBase IDs, or one of the other ID types listed. IDs should be separated by a new line.
- GENE ONTOLOGY: Restrict by one or more Gene Ontology (GO) terms for functional descriptions. Paste or upload a list of GO IDs or use the autocomplete box to populate the list.
Alternatively, you can restrict to a particular GO evidence type e.g. Inferred by Electronic Annotation (IEA). For an explanation of GO evidence codes see the GO website. Multiple codes can be selected by holding down the ctrl key, or option key on a Mac.
- HOMOLOGY: Restrict to orthologs or paralogs for a species by choosing from the pulldown list. You can choose to include (WITH) that ortholog/paralog set or to exclude (WITHOUT) that set from your query by using the checkboxes.
- PROTEIN DOMAINS: Allows you to restrict your query based on the presence or absence of protein domains.
Limit to genes...lets you choose a particular database feature set in include or exclude e.g. "restrict to all proteins containing any feature found in Pfam".
The second option, Limit to genes with these family or domain IDs:, allows you to restrict to one or more protein domains/families. Accepts IDs from several databases including InterPRO, Pfam and Panther. IDs should be separated by a new line.
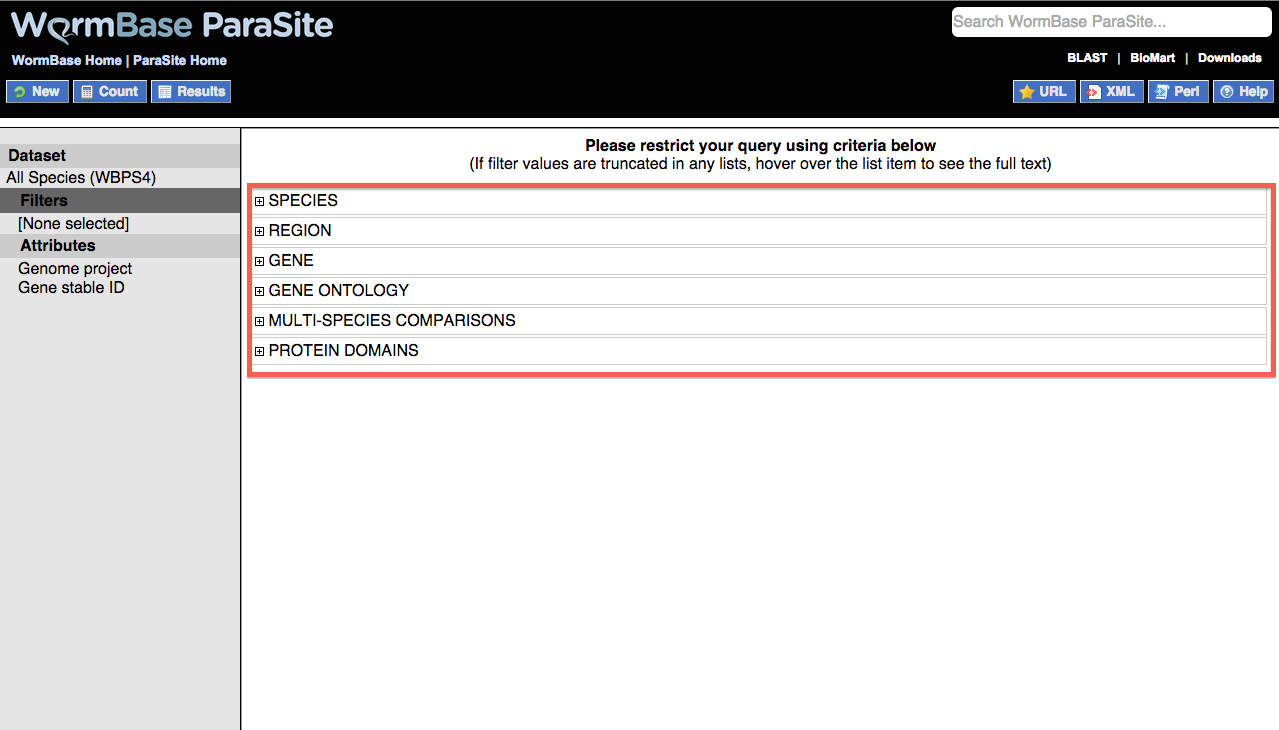
2) Setting your Attributes (outputs)
By default, BioMart is set to return a two-column list with genome assembly ID and gene ID e.g.
However, the Attributes field can be further configured to return additional columns of data. These types can broadly be divided into four groups:
Create a data table
Sequence output
- Unspliced (Transcript)
- Unspliced (Gene)
- Flank (Transcript)
- Flank (Gene)
- Flank-coding region (Transcript)
- Flank-coding region (Gene)
- 5' UTR
- 3' UTR
- Exon sequences
- cDNA sequences
- Coding sequence
- Peptide
By default this option is set to return genome project information (e.g. ascaris_suum_prjna62057) and gene ID as two columns.
Species information columns can also be included e.g. taxon ID and nematode clade.
Gene attributes that can be selected to appear in the output include gene, protein and transcript IDs, scaffold IDs, gene and transcript starts and ends, gene and transcript names and descriptions.
External references are cross-references to equivalent entities in other resources such as UniProt, RefSeq and ENA/GenBank as well as all WormBase IDs.
GO and GOslim options output various GO data including name, GO ID, definition and evidence code and nearest ancestor 'GO slim' term (see the GO slim guide for more information).
Protein domain adds columns for protein attributes such as InterPro domains and descriptions.
Exons in addition to Gene and Species attributes, this gives you output options for the exon structure of the gene such as CDS start and end and exon rank within the cDNA.
Homologues gives the option of exporting orthologs or homologs from one or more chosen species for a set of input genes. For example "given me all C. elegans orthologs for the B. malayi genes eat-3, eat-4 and eat-6, eat-16 and eat-20".
As well as the core Gene and Species attributes, additional output options include ortholog gene and protein IDs, chromosome/scaffold ID, orthology confidence and % identity.
A very useful feature of BioMart is the ability to output sequence in many different configurations:
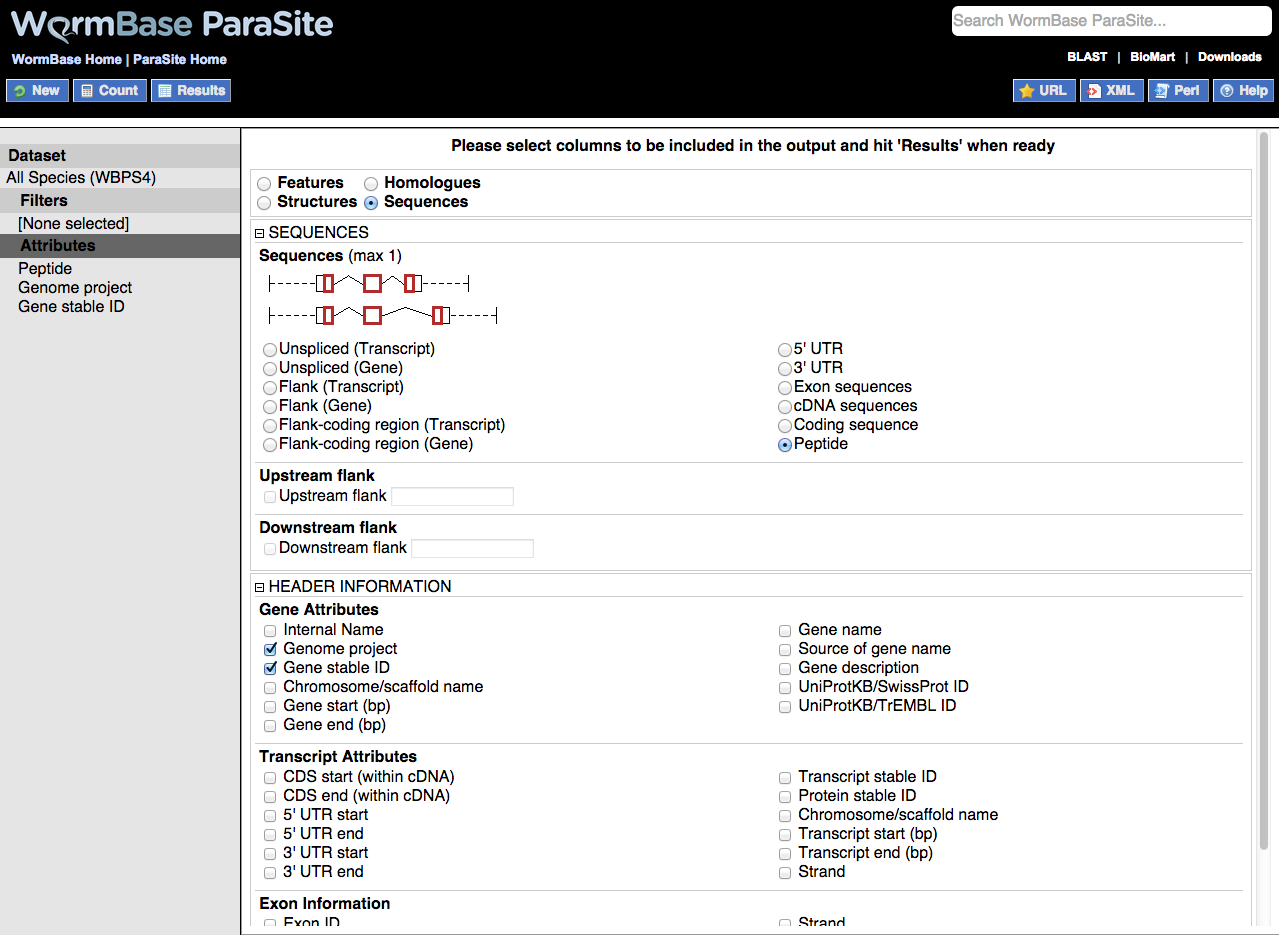
Output is a FASTA file where additional information can be given in the header lines. The sequences themselves can be generated using the following configurations. Only one option can be selected at a time.
Full transcript sequence, exons and introns. For genes with multiple transcripts, all transcript sequences are returned.
Full gene sequence, exons and introns.
Sequence adjacent to a transcript. Upstream flank or Downstream flank needs to be specified in the panel below, along with the length of the flanking sequence required in base pairs. For genes with multiple transcripts, all transcript flanking sequences are returned.
Sequence adjacent to a gene. Upstream flank or Downstream flank needs to be specified in the panel below, along with the length of the flanking sequence required in base pairs.
Flanking sequence for a transcript + 5' UTR sequence for upstream flank and 3' UTR sequence for downstream flank. Upstream flank or Downstream flank needs to be specified in the panel below, along with the length of the flanking sequence required in base pairs. For genes with multiple transcripts, all transcript sequences are returned.
Flanking sequence for a transcript + 5' UTR sequence for upstream flank and 3' UTR sequence for downstream flank. Upstream flank or Downstream flank needs to be specified in the panel below, along with the length of the flanking sequence required in base pairs. For genes with multiple transcripts, all transcript sequences are returned.
Sequence of the 5' untranslated region. Note that this sequence is only retrievable if the 5' UTR has been annotated for the gene(s) in question.
Sequence of the 3' untranslated region. Note that this sequence is only retrievable if the 3' UTR has been annotated for the gene(s) in question.
returns all exonic (coding) sequences. Each exon is returned as a separate sequence with its own header. Check 'Exon ID' in the Header section to get sequential numbering of exons within the gene/transcript.
All coding sequence including 5' and 3' UTRs.
All coding sequence excluding 5' and 3' UTRs.
Protein translation of the coding sequences. For genes with multiple transcripts, all transcript peptide sequences are returned.
Including additional information in sequence headers
Additional information can be added to sequences headers under the HEADER INFORMATION section. These extra columns include various gene and transcript attributes. Useful attributes to include are 'Gene name' and 'Gene description', and 'Transcript stable ID' which will allow you to differentiate transcripts where multiple transcripts exist for the same gene.
In addition, where exon sequence is being returned, several options are available under the Exon Information section for adding information to the headers of individual exon sequences. A useful thing to include would be 'Exon ID' which numbers the exons sequentially within the gene or transcript.
Once both the Filters and Attributes have both been set for your query, click the Resultsbutton to submit your query. The Count button gives you a preview of the number of results expected from the query. This can be useful to check if the size of your results is as you expect before running the full query.
3) Viewing and Downloading your Output
BioMart results can be viewed and downloaded from the results page.
Results are displayed on the page by default as a simple HTML table, and can also be shown as CSV (comma-separated values) or TSV (tab-separated values). For sequences the display format is FASTA.
For larger datasets the results can be exported as a file using the options at the top of the page. Export formats are HTML, CSV (comma-separated values), TSV (tab-separated values) and Excel (XLS). Sequences are exported as FASTA. For very large files, compressed formats are also available in the first pulldown menu.
4) Accessing data using R
The WormBase ParaSite BioMart fully supports the biomaRt package for R.
Install the biomaRt package:
source("http://bioconductor.org/biocLite.R")
biocLite("biomaRt")
Initialise biomaRt
library(biomaRt)
Establish a connection to the WormBase ParaSite BioMart
mart <- useMart("parasite_mart", dataset = "wbps_gene", host = "https://parasite.wormbase.org", port = 443)
List the available filters and attributes
listFilters(mart)
listAttributes(mart)
An example: get all the S. mansoni genes with a C. elegans orthologue
library(biomaRt)
mart <- useMart("parasite_mart", dataset = "wbps_gene", host = "https://parasite.wormbase.org", port = 443)
genes <- getBM(mart = mart,
filters = c("species_id_1010", "only_caelegprjna13758_homologue"),
value = list("scmansprjea36577", TRUE),
attributes = c("wbps_gene_id", "caelegprjna13758_gene", "caelegprjna13758_gene_name"))
head(genes)
Tip: to replicate a query constructed in the web browser, click the "XML" button. From here, you can see the relevant filter and attribute names required to construct the same query in biomaRt.

